FAQs
Can I account for spot distortions? Yes. SameSpots uses a next-generation pixel-level image alignment algorithm that warps gel images to remove the positional variation inherent to 2D electrophoresis. This includes distortions caused by gel-to-gel variation in running conditions, differences in gel casting, and imaging artefacts. The warping process aligns all images to a shared reference, ensuring that protein spots at different positions across gels are correctly matched before any quantitative analysis is performed.
Can I compare DIGE images? Yes. SameSpots fully supports 2D-DIGE analysis for experiments using Cy2, Cy3, and Cy5 fluorescent dyes, including experiments run with a pooled internal standard. The software automatically handles all channel management, normalization, and measurement calculations after alignment and co-detection, giving you statistically robust differential expression results with no missing values across all channels. DIGE support is included in the standard SameSpots license with no additional module purchase required.
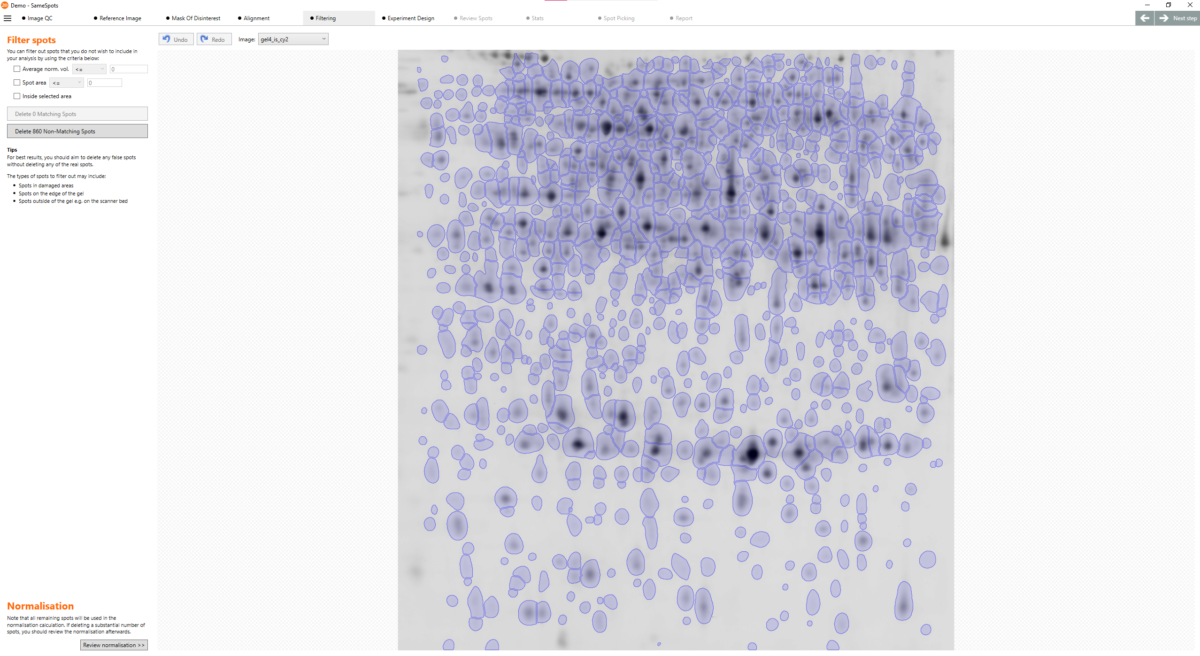
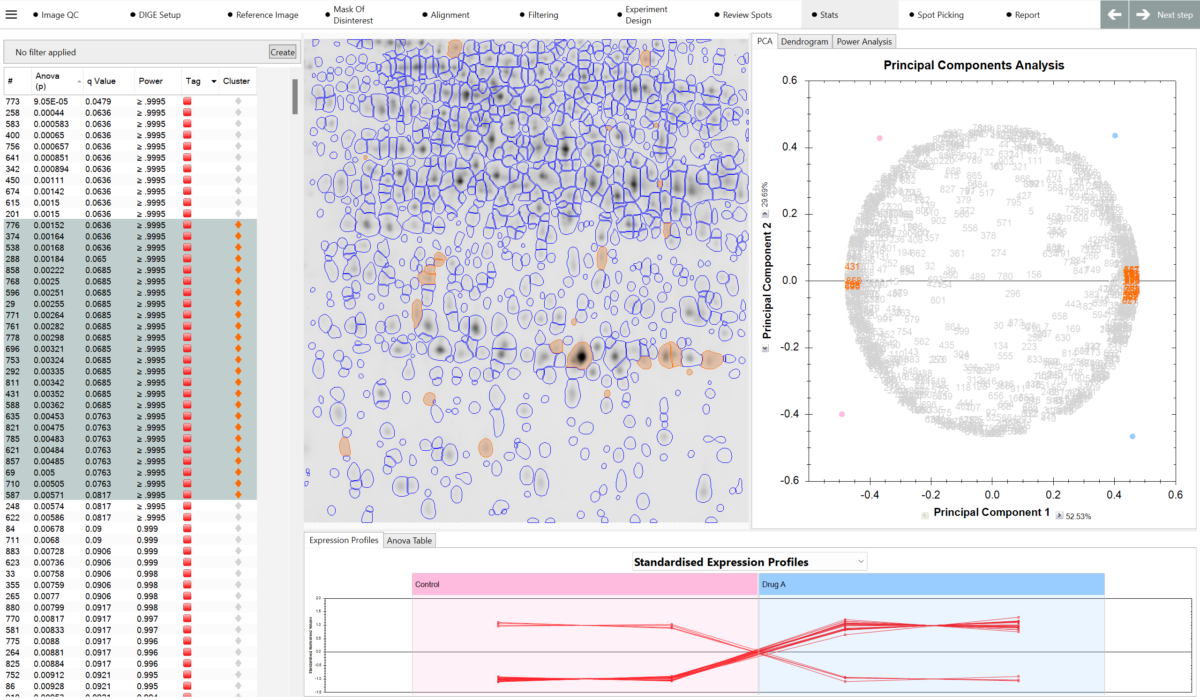
Can I match all spots across all images? Yes. SameSpots guarantees 100% spot matching with no missing values across all images in your experiment. Alignment removes positional variation between images, after which a single shared spot pattern is detected on a composite reference and automatically propagated to all images simultaneously. This means every spot is measured in every image, giving your statistical analysis a complete and unbroken dataset — a critical requirement for valid multivariate statistics in large proteomics experiments.
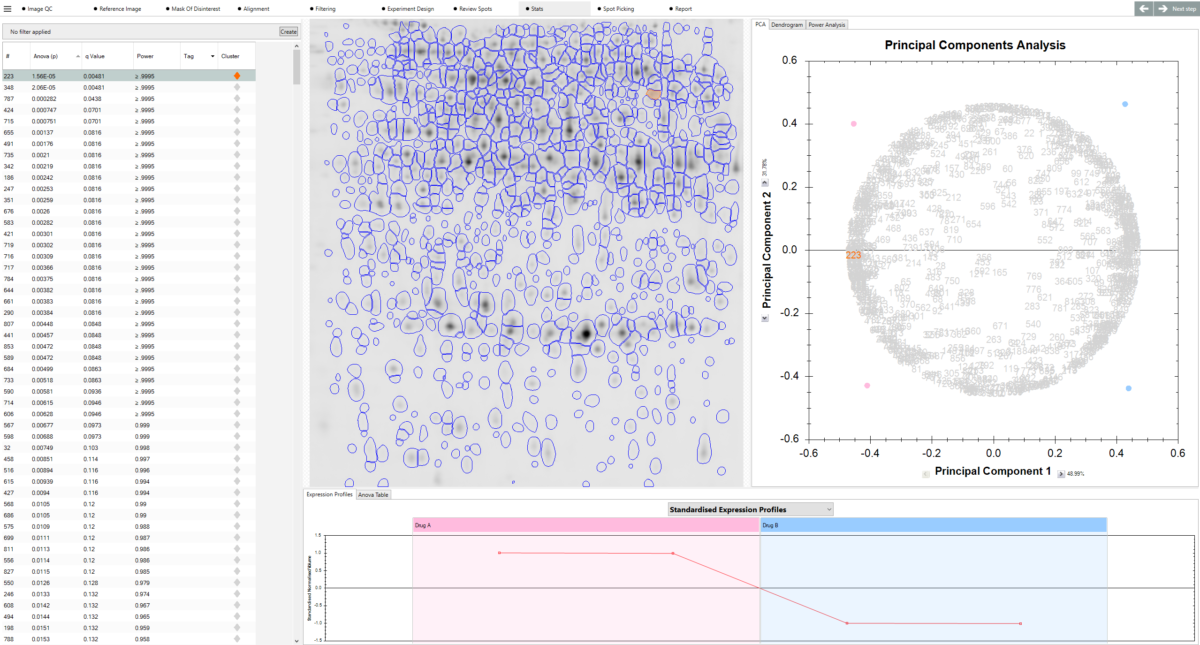
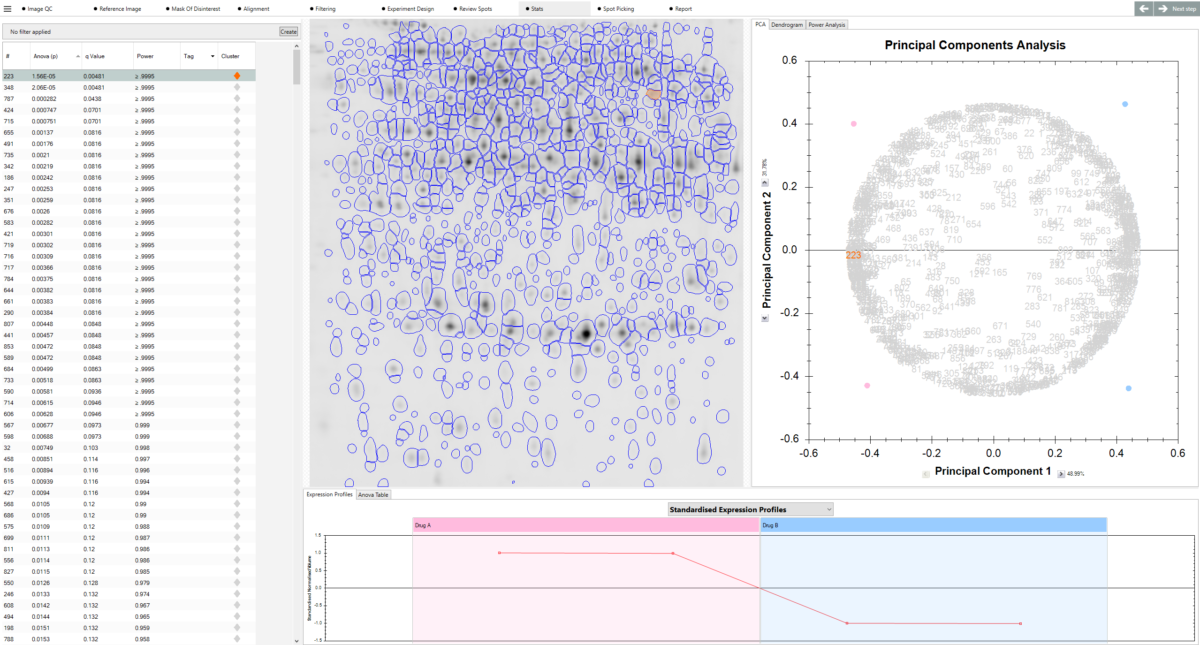
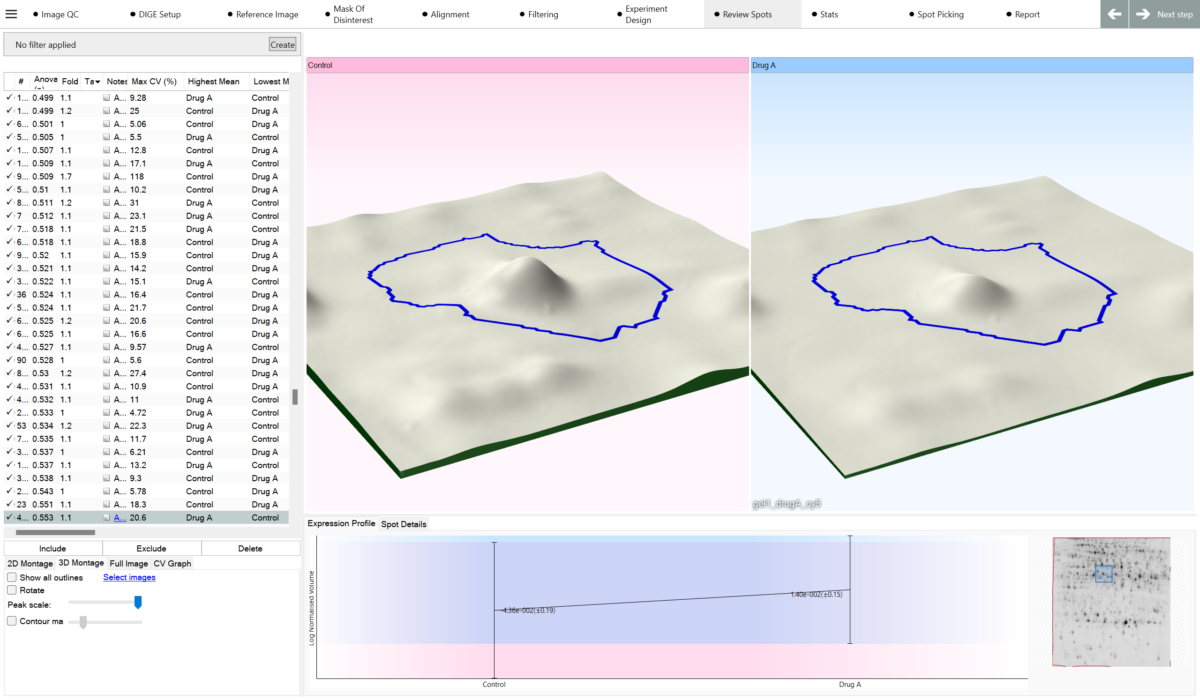
Can I highlight proteins of interest? Yes. SameSpots includes a quick-tagging system that allows you to flag protein spots showing significant changes in expression, making it easy to prioritise candidates for downstream analysis. Spots can be filtered and tagged based on statistical significance, fold change, spot abundance, or any combination of criteria. Tagged spots can then be exported directly to picking lists for mass spectrometry identification.
Can I export my results? Yes. SameSpots supports comprehensive data export at every stage of the analysis. You can export spot measurements, normalized abundances, statistical results, and analysis parameters to CSV for use in third-party software. The software also automatically generates a full PDF analysis report summarizing all QC checks, alignment results, detection parameters, statistical outputs, and images — suitable for inclusion in a LIMS, laboratory notebook, or regulatory submission.
What operating systems can I use? SameSpots is compatible with Windows 7, 8, 10, and 11. For regulated environments, SameSpots can be run in conjunction with TotalLab’s AuditSafe module to achieve full 21 CFR Part 11 and EU Annex 11 compliance on supported Windows operating systems.
What can I use to decide which spots are interesting? SameSpots provides a full suite of analytical tools to help you identify proteins of interest, including one-way ANOVA, two-way ANOVA, repeated measures ANOVA, principal component analysis (PCA), expression profiles, interaction plots, scatter plots, and descriptive statistics including fold change, mean, standard deviation, and coefficient of variation. Statistical tests are automatically selected and configured based on your experimental design. You can combine statistical filters with abundance thresholds and manual tagging to build a shortlist of candidates with confidence.
Can I check my gel running quality? Yes. SameSpots includes image quality control tools that flag potential issues with your gel images before you invest time in analysis. Quality checks include assessment of bit-depth, image file size, saturation, and consistency of image characteristics across the dataset. Any issues are highlighted immediately upon image import, along with guidance on how to address them — helping you avoid running a full analysis on images that are unlikely to deliver reliable results.
Can I check my image quality? Yes. When adding images to the experiment they are automatically checked for quality and any issues are highlighted immediately before any further analysis is attempted. SameSpots checks for saturation, bit-depth suitability, image size consistency, and other parameters that affect the reliability of spot detection and quantification. Addressing image quality issues at this stage saves significant time compared to discovering problems after a full analysis has been run.
What checks does Image QC perform? SameSpots Image QC checks that images meet the requirements for reliable automated analysis, including: bit-depth (16-bit images are strongly recommended for quantitative analysis), saturation (overexposed spots cannot be accurately quantified), image resolution and file size consistency across the dataset, and image inversion or orientation issues. Where issues are detected, the software provides specific guidance on corrective action — for example, re-scanning at a lower sensitivity to eliminate saturated spots.
Can I edit my images? Yes. SameSpots includes image editing tools for cropping, rotating, flipping, scaling, and inverting images. Basic editing operations that are applied in the context of a DIGE experiment are automatically applied simultaneously to all images from the same gel, maintaining consistency across channels. Spot editing tools allow you to add, remove, split, merge, grow, shrink, or move spots after automated detection, with all edits propagated simultaneously across all images to maintain 100% spot matching throughout.
How can I account for different gel loading? Variation in sample loading between lanes or gels is corrected through normalization. SameSpots supports multiple normalization approaches including the default ratiometric method, total volume normalization, and spike normalization using housekeeping proteins or exogenous spike proteins. Normalization is applied before statistical analysis and is reviewed in a dedicated step in the workflow, allowing you to verify that the normalization approach is appropriate for your experimental design before proceeding to results.
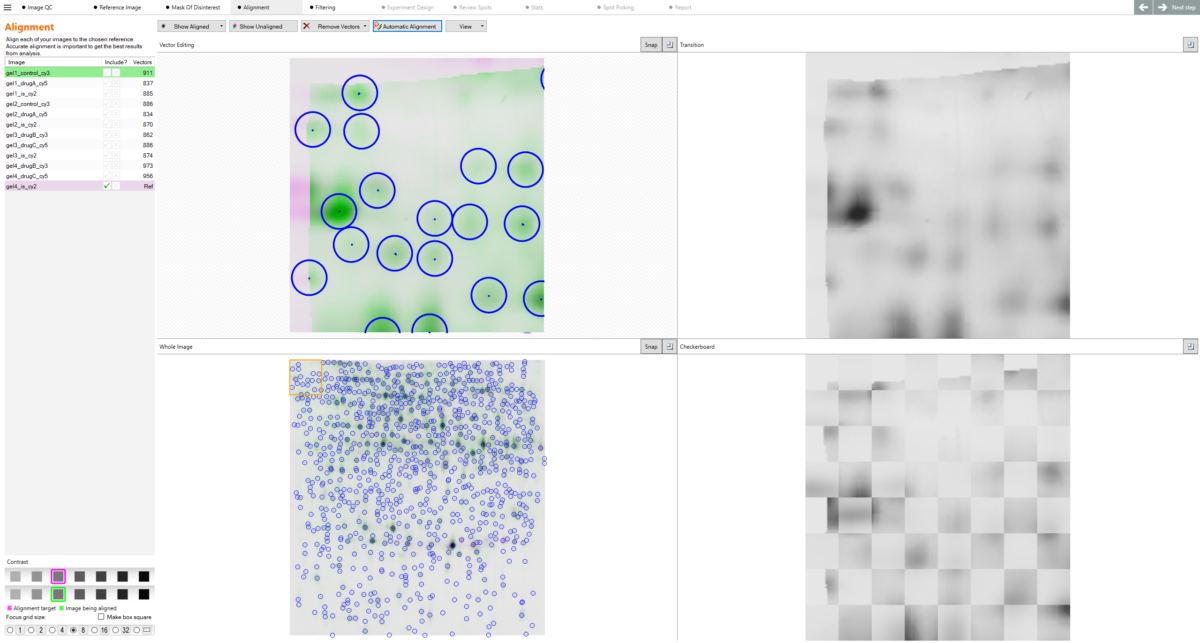
What is alignment and what problem does it solve? Alignment is the process of removing the positional variation that is inherent to 2D gel electrophoresis. Because no two gels are physically identical — even when run under identical conditions — the same protein spot will appear at a slightly different position on each gel. Without alignment, comparing spot intensities across gels would be unreliable, since you cannot be certain you are measuring the same protein in each image. SameSpots’ alignment algorithm warps each image so that all protein spots are brought into the same coordinate space, making accurate cross-gel comparison possible.
What is the reference gel? The reference gel is the image that all other images in the experiment are aligned to. SameSpots automatically selects the most suitable reference image based on image quality and spot pattern representativeness, though you can change this selection if preferred. A good reference image should have a large number of clearly resolved, representative spots. Once alignment is complete, a composite reference is generated from all aligned images, and the spot pattern is detected on this composite rather than on the reference gel alone — ensuring a more representative and complete spot map.
How does alignment work? SameSpots alignment operates by identifying corresponding spots between each image and the reference image, then warping the image so that its spots precisely superimpose with those in the reference coordinate space. This is performed automatically using TotalLab’s next-generation pixel-level alignment algorithms, which are designed to handle the full range of positional variation encountered in real experimental datasets. After automatic alignment, you can review each alignment pair using side-by-side, dual-color, or blink display modes and make manual corrections where needed using 2D and 3D editing views.
Can I calculate pI and MW? Yes. SameSpots supports pI and MW calibration by allowing you to define known pI and MW markers within your gel images. Once defined, the software calculates theoretical pI and MW values for all detected spots, which can be used to aid protein identification and to cross-reference results with mass spectrometry data or protein databases such as MASCOT.
Can I import mass spectrometry data? Yes. Protein identifications from mass spectrometry can be imported back into SameSpots from standard proteomics databases including MASCOT, as well as from custom databases. Once imported, protein identifications are displayed alongside the corresponding spot quantitation data in the analysis and are included in the exported analysis report, giving you a complete and integrated view of expression changes and protein identity in a single document.






















 Powered by Bioz
Powered by Bioz